
hATTR amyloïdose ontrafelt hun leven
Als hATTR amyloïdose in een vroeg stadium wordt herkend en behandeld, kan dit patiënten helpen om hun fysieke functie, onafhankelijkheid en welzijn te behouden.1,2,7,10
Wat is hATTR-amyloïdose?
Erfelijke ATTR-amyloïdose (hATTR-amyloïdose) is een progressieve, multisysteemziekte die het gevolg is van de ophoping van gemuteerd TTR-eiwit – voornamelijk geproduceerd door de lever – in de zenuwen, het hart en andere organen. Het kan onder andere zenuwproblemen op meerdere plaatsen in het lichaam (polyneuropathie) veroorzaken.1,2
Erfelijke ATTR-amyloïdose wordt veroorzaakt door een mutatie van het TTR-gen. De mutatie zorgt ervoor dat het gevormde TTR-eiwit onstabiel wordt. Dit instabiele TTR valt uit elkaar in kleine stukjes, die zich extracellulair opstapelen tot amyloïde-afzettingen in verschillende organen en weefsels, zoals de zenuwen, het hart en de nieren.1

Ontwikkeld door Alnylam Pharmaceuticals
De impact van misdiagnose
De vroegtijdige symptomen van erfelijke ATTR-amyloïdose worden vaak over het hoofd gezien.3-5
hATTR-amyloïdose is een zeer zeldzame ziekte met een complex diagnose-traject. De symptomen kunnen zich namelijk op veel verschillende vlakken manifesteren. Patiënten krijgen daarom helaas vaak pas na jaren de diagnose hATTR-amyloïdose, waardoor hun kwaliteit van leven en overlevingskans verslechteren.
Het stellen van de juiste diagnose duurt gemiddeld 3 tot 4 jaar.6
Een vroege diagnose kan het verloop van de ziekte veranderen en voorkomt onomkeerbare schade.2,5,7-10
Het is daarom het belangrijk snel te (be)handelen. Behandeling kan de achteruitgang van de ziekte vertragen of zelfs stoppen en zelfs enige functie herstellen. Hiervoor is het vroegtijdig ingrijpen van cruciaal belang.1,2,7,10
Hoe herken je hATTR-amyloïdose?
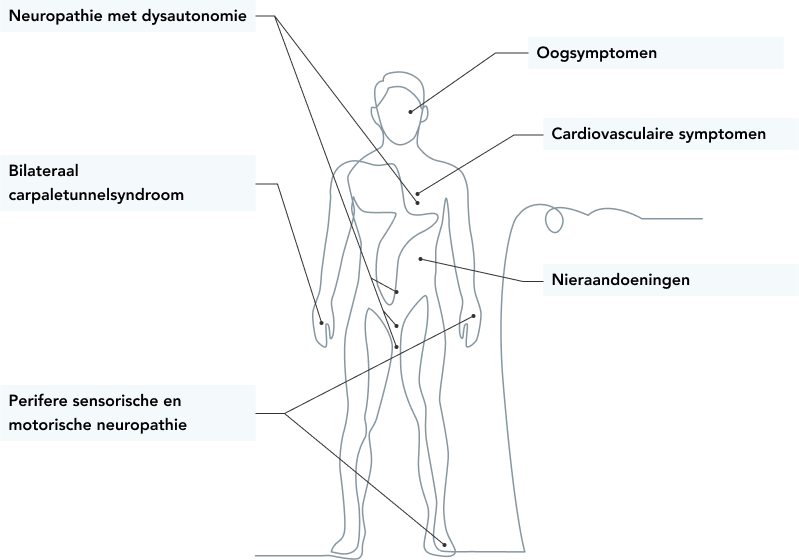
Erfelijke ATTR-amyloïdose kan op verschillende manieren tot uiting komen.1,11,12
Patiënten met hATTR-amyloïdose kunnen symptomen vertonen over een breed spectrum, waaronder perifere sensomotorische neuropathie, autonome disfunctie en cardiomyopathie.1,12
Omdat de symptomen dusdanig divers zijn, worden patiënten die mogelijk lijden aan hATTR-amyloïdose vaak al vanuit meerdere disciplines behandeld (neurologie, cardiologie, interne geneeskunde of bijvoorbeeld pijnpoli). Dit kan de prognose negatief beïnvloeden, omdat erfelijke ATTR-amyloïdose vaak niet tijdig wordt gediagnosticeerd.1,13
Erfelijke ATTR-amyloïdose kan een verwoestend effect hebben op het leven van patiënten.13,14
Het ontneemt hun onafhankelijkheid en vermindert de kwaliteit van leven.13-14
Zonder behandeling kan de ziekte zich snel en meedogenloos ontwikkelen, zelfs met de dood – binnen enkele jaren – tot gevolg.1,2,13,15,16
Klinische bevindingen die kunnen wijzen op erfelijke ATTR amyloïdose

Voorgeschiedenis en klinische symptomen
- Hartfalen met een normale of behouden ejectiefractie in afwezigheid van hypertensie, vooral bij mannen
- Hypotensie bij een persoon met een voorgeschiedenis van hypertensie
- Syncope, geleidingsstoornissen
- Intolerantie voor veel gebruikte cardiovasculaire geneesmiddelen: remmers van angiotensineconverterend enzym, angiotensinereceptorblokkers en bètablokkers

Cardiale beeldvorming
ECG
- Microvoltage
- Pseudo-aspect – Q-golf
- Atrioventriculaire geleidingsstoornis
Echografie
- Concentrische linkerventrikelhypertrofie > asymmetrisch
- Afwijkende strain van het linkerventrikel (basaal >apicaal)
- Verdikking van de (mitrale en tricuspide) kleppen
- Pericardiale effusie
- Rechterventrikelhypertrofie (>5mm)
Cardiale MRI
- Morfologische afwijkingen (zie echo)
Scintigrafie-onderzoek
- Cardiale fixatie van de tracer 99mTc-DPD of 99mTc-PYP voor botscintigrafie
Denk aan hATTR amyloïdose bij een patiënt met een familiale voorgeschiedenis en/of een van deze symptomen.
ECG=elektrocardiogram; cardiale MRI=cardiale magnetische resonantie beeldvorming; 99mTc-DPD=technetium-99m-3,3-difosfono-1,2-propaan-dicarbonzuur; 99mTc-PYP=technetium -99m-pyrofosfaat. Aangepast van Dharmarajan K, Maurer M. J Am Geriatr Soc. 2012;60(4):765-774.
Evolutieve neuropathie met minstens 1 van de volgende elementen:

Dysautonomie
- Erectiele disfunctie
- Orthostatische hypotensie
- Gastro-intestinale stoornissen (diarree, constipatie of afwisseling diarree/constipatie)
Loop- en/of gevoelsstoornissen
Onverklaarbaar gewichtsverlies
Bilateraal carpaletunnel-syndroom
Familiale voorgeschiedenis van amyloïdose
Cardiovasculaire symptomen
bv. geleidingsblok, cardiomyopathie, of aritmie)
Glasvochttroebelingen
Nefropathie
(bv. proteïnurie en nierinsufficiëntie)
Bijkomende tekenen: snelle ziekteprogressie en falen van immunomodulerende behandeling.
Klinische bevindingen die kunnen wijzen op erfelijke ATTR amyloïdose
Voorgeschiedenis en klinische symptomen
- Hartfalen met een normale of behouden ejectiefractie in afwezigheid van hypertensie, vooral bij mannen
- Hypotensie bij een persoon met een voorgeschiedenis van hypertensie
- Syncope, geleidingsstoornissen
- Intolerantie voor veel gebruikte cardiovasculaire geneesmiddelen: remmers van angiotensineconverterend enzym, angiotensinereceptorblokkers en bètablokkers
Cardiale beeldvorming
ECG
- Microvoltage
- Pseudo-aspect – Q-golf
- Atrioventriculaire geleidingsstoornis
Echografie
- Concentrische linkerventrikelhypertrofie > asymmetrisch
- Afwijkende strain van het linkerventrikel (basaal >apicaal)
- Verdikking van de (mitrale en tricuspide) kleppen
- Pericardiale effusie
- Rechterventrikelhypertrofie (>5mm)
Cardiale MRI
- Morfologische afwijkingen (zie echo)
Scintigrafie-onderzoek
- Cardiale fixatie van de tracer 99mTc-DPD of 99mTc-PYP voor botscintigrafie
Denk aan hATTR amyloïdose bij een patiënt met een familiale voorgeschiedenis en/of een van deze symptomen.
ECG=elektrocardiogram; cardiale MRI=cardiale magnetische resonantie beeldvorming; 99mTc-DPD=technetium-99m-3,3-difosfono-1,2-propaan-dicarbonzuur; 99mTc-PYP=technetium -99m-pyrofosfaat. Aangepast van Dharmarajan K, Maurer M. J Am Geriatr Soc. 2012;60(4):765-774.
Evolutieve neuropathie met minstens 1 van de volgende elementen:
Dysautonomie
- Erectiele disfunctie
- Orthostatische hypotensie
- Gastro-intestinale stoornissen (diarree, constipatie of afwisseling diarree/constipatie)
Loop- en/of gevoelsstoornissen
Onverklaarbaar gewichtsverlies
Bilateraal carpaletunnel-syndroom
Familiale voorgeschiedenis van amyloïdose
Cardiovasculaire symptomen
bv. geleidingsblok, cardiomyopathie, of aritmie)
Glasvochttroebelingen
Nefropathie
(bv. proteïnurie en nierinsufficiëntie)
Bijkomende tekenen: snelle ziekteprogressie en falen van immunomodulerende behandeling.
Hulp bij diagnosticeren
Het is van levensbelang dat patiënten met hATTR-amyloïdose tijdig worden gediagnosticeerd. Echter, het is niet altijd eenvoudig om de diagnose hATTR-amyloïdose te stellen in geval van polyneuropathische klachten. Of om onderscheid te maken met CIDP, vanwege de overlap aan symptomen.5,18,19
Er zijn op basis van studies een aantal ‘rode vlaggen’ gesignaleerd; klinische signalen die het overwegen van de diagnose hATTR-amyloïdose, verder onderzoek en eventueel bijbehorend genetisch onderzoek rechtvaardigen.5,18,19 De rode vlaggen hebben we verzameld in een handig overzicht dat u kunt downloaden.
Herken de signalen en symptomen van hATTR-amyloïdose vanuit uw specialisme en bij vermoeden, bespreek de patiënt tijdens het MD-overleg.5,18,19

Wanneer u een patiënt met hartklachten (o.a. hartfalen) tegenkomt die tevens symptomen van polyneuropathie vertoont, is het belangrijk om waakzaam te zijn. U kunt het complete overzicht van alle ‘rode vlaggen’ cardiologie hieronder downloaden.

Wees extra alert wanneer u een patiënt tegenkomt met een evolutieve polyneuropathie in combinatie met dysautonomie, loop- en/of gevoelsstoornissen, gewichtsverlies, bilateraal carpaal tunnel syndroom of cardiovasculaire symptomen. Of als de patiënt niet goed reageert op de behandeling van CIDP.⁵,¹⁸,¹⁹ Lees alles wat u moet weten over de ‘rode vlaggen’ vanuit het expertisegebied neurologie in het handige overzicht hieronder.
CIPD als misdiagnose
Tussen 15 en 20% van de hATTR amyloïdose patiënten werd eerder gediagnosticeerd met CIDP⁴⁻⁶
Geregeld denken artsen bij een sensomotorische neuropathie aan de zeldzame aandoening ‘chronische inflammatoire demyeliniserende polyneuropathie’ (CIDP).20 Bij een deel van de gevallen met CIDP is sprake van een misdiagnose21 en blijken de symptomen te berusten op een erfelijke vorm van ATTR-amyloïdose (hATTR).5
Erfelijke of wild-type ATTR?
Doe steeds de genetische test
Om de juiste diagnose te stellen is het belangrijk om te weten om welk type van ATTR-amyloïdose het gaat. Het is daarom belangrijk om altijd een genetische test te laten uitvoeren. De genetische ziekte hATTR is namelijk sneller progressief dan het wild-type en vraagt steeds een multidisciplinaire zorg. Bovendien is de kennis van het bestaan van een genetische ziekte belangrijk voor de andere leden van de familie.
| Verschillen tussen hATTR en wtATTR | |||
|---|---|---|---|
Erfelijke ATTR amyloïdose (hATTR) | Wild Type ATTR amyloïdose (wtATTR) | ||
Genetisch²⁶,²⁷ Typische leeftijd bij presentatie²⁷,²⁸ | Mutatie in het TTR gen 25-65 jaar (afhankelijk van de mutatie) | Geen genetische mutatie in het TTR-gen > 60-70 jaar | |
Soorten manifestaties²⁷,²⁸ |
| ||
|
| ||
Prognose bij afwezigheid van behandeling | ~ 2 jaar* | ~ 4 jaar* | |
Erfelijke ATTR amyloïdose heeft een ander verloop dan Wild type ATTR amyloïdose, met een kortere overleving.²⁹,³⁰
*Patiënten die zich voornamelijk met hartklachten presenteren.

Consensusaanbeveling voor diagnose van ATTR amyloïdose met polyneuropathie
* U staat op het punt een website te bekijken die niet wordt beheerd door Alnylam Pharmaceuticals, en het bedrijf bevestigt de inhoud en nauwkeurigheid ervan of andere praktijken of normen die worden vermeld, niet. Alle handelsmerken zijn het eigendom van hun respectieve eigenaren.
De enige manier om zekerheid te krijgen over de diagnose is met een genetisch onderzoek³
Als er een klinisch vermoeden is van cardiale ATTR amyloïdose, raden de meest recente richtlijnen scintigrafie of biopsie aan om TTR amyloïdose te identificeren, gevolgd door een genetisch onderzoek om erfelijke of wild-type amyloïdose te bevestigen³
* U staat op het punt een website te bekijken die niet wordt beheerd door Alnylam Pharmaceuticals, en het bedrijf bevestigt de inhoud en nauwkeurigheid ervan of andere praktijken of normen die worden vermeld, niet. Alle handelsmerken zijn het eigendom van hun respectieve eigenaren.
Heeft uw patiënt hATTR amyloïdose? Verwijs door naar een Neuro Musculair Referentie Centrum (NMRC)
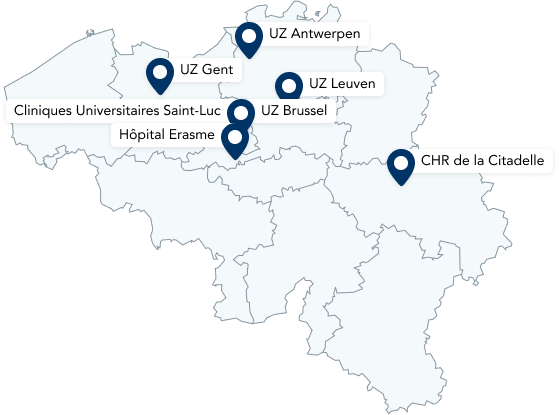
Het expertise centrum voor hATTR is een Neuro Musculair Referentie Centrum (NMRC)
Wanneer u erfelijke amyloïdose diagnosticeert bij uw patiënt, is het aangeraden om hem door te verwijzen naar een erkend neuromusculair referentiecentrum (NMRC) in uw buurt. De expert neuroloog zal aldaar de patiënt onderzoeken op vlak van polyneuropathie en de nodige multidiscipliniare zorg opstarten.
In België wordt deze zorg geboden in de expertcentra voor erfelijke ATTR amyloïdose, de Neuro-Musculaire Referentie Centra (NMRC). In het NMRC zijn er additionele medicamenteuze mogelijkheden, zoals behandeling met RNAi-medicatie. U vindt een NMRC in elk universitair ziekenhuis.
Bent u geïnteresseerd in een gesprek met een Alnylam-vertegenwoordiger?
Om meer te weten te komen over onze producten en/of therapeutische gebieden, klik op de knop ‘Connect with Alnylam’of stuur een e-mail naar info@alnylam.be
Heeft u een patiënt met amyloïdose?
Informeer hem over de Belgische patiëntenvereniging voor patiënten met amyloïdose.

Afkortingen:
- ATTR: transthyretine amyloïdose
- TTR: transthyretine
- CIPD: chronisch inflammatoire demyeliniserende polyneuropathie
- NMRC: neuromusculair referentiecentrum
- RNA: ribonucleic acid
Referenties:
- Adams D, et al. Nat Rev Neurol. 2019;15(7):387–404
- Kristen AV, et al. Neurodegener Dis Manag. 2019;9(1):5–23
- Grogan M, et al. Presented at 23rd Annual Scientific Meeting of Heart Failure Society of America (HFSA); Philadelphia PA, USA; September 13–16, 2019. Poster
- Lane T, et al. Circulation. 2019;140:16–26
- Gertz M, et al. BMC Fam Pract. 2020;21:198
- Adams D, et al. J Neurol. 2020; doi: 10.1007/s00415-019-09688-0
- Adams D, et al. Lancet Neurol. 2020;doi.org/10.1016/S1474-4422(20)30368-9
- Brannagan TH, et al. Eur J Neurol. 2020;27:1374–1381
- Schmidt HH, et al. Muscle Nerve. 2016;54(3):353–360
- Benson MD, et al. Ther Clin Risk Manag. 2020;16:749–758
- Ruberg FL, et al. J Am Coll Cardiol. 2019;73(22):2872–2891
- Waddington Cruz M, et al. Orphanet J Rare Dis. 2019;14:34.
- González-Duarte A, et al. J Neurol. 2020;267(3):703–712.
- Obici L, et al. Amyloid. 2020;27(3):153–162.
- Swiecicki PL, et al. Amyloid. 2015;22(2):123–131.
- Sattianayagam PT, et al. Eur Heart J. 2012;33(9):1120–1127.
- Gonzalez-Duarte A, et al. Clin Auto Res. 2019;29(Suppl 1):S1–S9.
- Adams D, et al. J Neurol. 2021;268:2109–22
- Hurtevent A, et al. Poster of final results.
- Van den Bergh PYK, et al. J Peripher Nerv Syst 2021 Sep;26(3):242-268.
- Lehmann HC, et al. J Neurol Neurosurg Psychiatry 2019 Sep;90(9):981-987.
- Vyndaqel European Public Assessment Report, 12 December 2019
- Kittleson MM, et al. Circulation. 2020;142(1):e7–e22.
- Maurer MS, et al. Circ Heart Fail. 2019;12(9):e006075.
TTR-BELUX-00057 mei 2025